


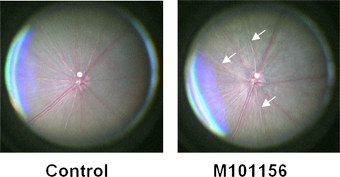
A model for retinitis pigmentosa (RP)B6(D2)-Rom1Rgsc1156 (M101156) RBRC GSC 0298
|
|
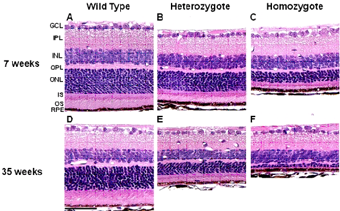 Figure 2. Light photomicrographs of the retina from each genotype of M-1156 mice (Rom1+/+, Rom1Rgsc1156/+, and Rom1Rgsc1156/Rgsc1156) at 7 (A, A, C) and 35 (D, E, F) weeks old. The outer nuclear layer (ONL) was thinner, and the inner and outer segments (IS and OS) of the photoreceptors were shorter in heterozygotes (B, E) than in wild-type mice (A, D). Additionally, the ONL in homozygotes (C, F) was thinner than that in heterozygotes (B, E). These alterations were more pronounced at 35 weeks of age than at 7 weeks of age. |
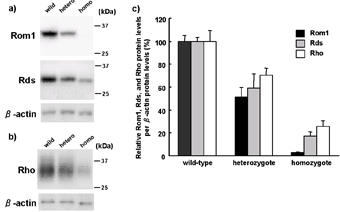 Figure 3. Western blot analysis of Rom1, Peripherin/Rds, and Rhodopsin proteins of the retina from wild-type (N = 6), heterozygote (N = 6), and homozygote (N = 6) mice at 3 weeks old. a) Western blotting of 10 μg protein from the retina with Rom1, Rds, and β-actin antibody. b) Western blotting of 0.1μg protein from retina with Rho and β-actin antibody. c) Semi-quantitative analysis of Rom1, Rds, and Rho protein levels by genotype. Protein levels in each gene showed significant differences by genotype (one-way ANOVA, P < 0.001). Post hoc Tukey Kramer multiple comparisons test revealed that the protein levels were significantly decreased by genotype in all the three genes (P < 0.05). |
Retinitis pigmentosa (RP) is a genetically heterogeneous disorder. RP patients progressively lose vision due to the gradual death of both rods and cones throughout the retina. Rod outer segment membrane protein 1 (Rom1) and peripherin/rds (Prph2) are localized along the rim region of the photoreceptor discs. rds mice that are homozygous for a null mutation in peripherin/rds (Prph2) fail to develop outer segments, whereas heterozygous rds mice exhibit formation of highly disorganized structures. On the other hand, homozygous Rom1 knockout mice develop outer segments with only mild defects. Mutations in peripherin/retinal degeneration, slow (PRPH2) cause hereditary human retinal degenerative diseases that are characterized by autosomal dominant RP and macular dystrophy. In contrast, there is no clear evidence to date of mutations in retinal outer segment membrane protein 1 (ROM1) alone causing retinal degeneration. Although it is well known that only persons who are double heterozygous for ROM1 and PRPH2 develop RP in a digenic inheritance pattern.
A monogenic dominant mutation in Rom1 of B6(D2)-Rom1Rgsc1156 causes retinal degeneration. The results suggest that Trp182Arg substitution of Rom1 decreases the levels of wild-type Prph2 and Rom1, which in turn causes a reduction in the level of Prph2-containing tetramers in the disc rim region, which may ultimately result in unstable and disorganized outer segments and photoreceptor degeneration.
A monogenic dominant mutation in Rom1 of B6(D2)-Rom1Rgsc1156 causes retinal degeneration. The results suggest that Trp182Arg substitution of Rom1 decreases the levels of wild-type Prph2 and Rom1, which in turn causes a reduction in the level of Prph2-containing tetramers in the disc rim region, which may ultimately result in unstable and disorganized outer segments and photoreceptor degeneration.
| Depositor | : | Mouse Functional Genomics Research Group, RIKEN Genomic Sciences Center |
| Reference | : | Sato H et al. A monogenic dominant mutation in Rom1 generated by N-ethyl-N-nitrosourea mutagenesis causes retinal degeneration in mice. Mol. Vision 16, 378-391, 2010 |

