


Targeted disruption of exon 52 in the dystrophin geneB6.129-Dmd<tm1Mok>/NahRbrc (RBRC03556)
Courtesy of Madoka Uezumi-Ikemoto, Ph.D. and Naohiro Hashimoto, Ph.D. Tibialis anterior muscles of 5-week-old male C57BL/6 (left panels) and C57BL/6 background mdx52 (right panels) mice. Upper panels, dystrophin was recognized by the NCL-DYS2 antibody (green) and nuclei were stained with DAPI (blue). Lower panels, sections were stained with hematoxylin and eosin. Scale bar, 100 µm. |
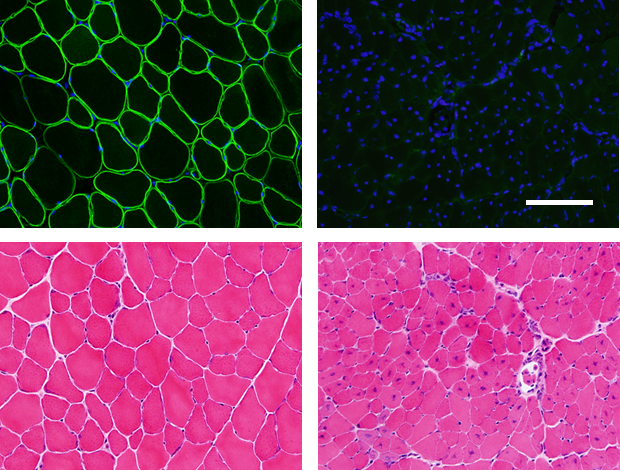
| Muscular dystrophies are progressive muscle disorders and are characterized by muscle fiber degeneration and necrosis. Duchenne muscular dystrophy (DMD), a lethal, X-linked disease that occurs in 1 out of every 3,500 male births, is the most common type of muscular dystrophy. DMD is caused by a mutation in the dystrophin gene, which is one of the largest genes in the human genome. Frameshift deletions, nonsense mutations, and other mutations in the dystrophin gene result in dystrophin loss, muscle fiber necrosis, and progressive replacement of muscle fiber by fibrotic tissue and fat. |
| Various mouse models with mutations in the mouse dystrophin gene have been reported, such as mdx mice [1]. Mdx mice have a spontaneous nonsense point mutation in exon 23 and lack dystrophin expression. Katsuki and colleagues generated mdx52 mice, which have a deletion mutation of exon 52 [2]. Mdx52 mice lack dystrophin and exhibit dystrophic changes and muscle hypertrophy [3]. A recent report suggested that mutation type and aging differentially affect muscle regeneration and proliferation in mdx and mdx52 mice with the same C57BL/6 background [4]. |
| One possible therapy for DMD has been designed to skip an exon of the dystrophin gene in order to induce an in-frame mutation and make a shorter but nevertheless functional form of the dystrophin protein. Exon skipping has been performed in mdx52 mice and a DMD canine model using antisense oligonucleotides, such as phosphorodiamidate morpholino oligomers (PMOs). The results of these studies show that administration of PMOs restored the reading frame and induced the expression of partially functional dystrophin [5-7]. This C57BL/6J congenic strain of the mdx52 mouse was developed in 2007 by Dr. Hashimoto at National Center for Geriatrics and Gerontology and is useful for developing therapies for DMD. |
| Depositor | : | Naohiro Hashimoto, Ph.D. Department of Regenerative Medicine National Center for Geriatrics and Gerontology |
|
| Strain name | : | B6.129-Dmd<tm1Mok>/NahRbrc | |
| RBRC No. | : | RBRC03556 | |
| Model to human disease | : | MUSCULAR DYSTROPHY, DUCHENNE TYPE; DMD OMIM ID: 310200 |
|
| Related resources from RIKEN BRC | : | Disease-specific iPS cells (patient-specific iPS cells) Muscular dystrophy, Duchenne type |
|
| References | : | [1] | Bulfield G, Siller WG, Wight PA, Moore KJ. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc Natl Acad Sci U S A.;81(4):1189-92, 1984. |
| [2] | Araki E, Nakamura K, Nakao K, Kameya S, Kobayashi O, Nonaka I, Kobayashi T, Katsuki M. Targeted disruption of exon 52 in the mouse dystrophin gene induced muscle degeneration similar to that observed in Duchenne muscular dystrophy. Biochem Biophys Res Commun.; 238(2):492-7, 1997. | ||
| [3] | Nakamura A, Takeda S. Mammalian models of Duchenne Muscular Dystrophy: pathological characteristics and therapeutic applications. J Biomed Biotechnol.; 2011:184393, 2011. | ||
| [4] | Echigoya Y, Lee J, Rodrigues M, Nagata T, Tanihata J, Nozohourmehrabad A, Panesar D, Miskew B, Aoki Y, Yokota T. Mutation types and aging differently affect revertant fiber expansion in dystrophic mdx and mdx52 mice. PLoS One.; 8(7):e69194, 2013. | ||
| [5] | Yokota T, Lu QL, Partridge T, Kobayashi M, Nakamura A, Takeda S, Hoffman E. Efficacy of systemic morpholino exon-skipping in Duchenne dystrophy dogs. Ann Neurol.; 65(6):667-76, 2009. | ||
| [6] | Aoki Y, Nakamura A, Yokota T, Saito T, Okazawa H, Nagata T, Takeda S. In-frame dystrophin following exon 51-skipping improves muscle pathology and function in the exon 52-deficient mdx mouse. Mol Ther.; 18(11):1995-2005, 2010. | ||
| [7] | Aoki Y, Yokota T, Nagata T, Nakamura A, Tanihata J, Saito T, Duguez SM, Nagaraju K, Hoffman EP, Partridge T, Takeda S. Bodywide skipping of exons 45-55 in dystrophic mdx52 mice by systemic antisense delivery. Proc Natl Acad Sci U S A.; 109(34):13763-8, 2012. | ||
| April 2014 Contact: Shinya Ayabe, Ph.D. Experimental Animal Division, RIKEN BioResource Center All materials contained on this site may not be reproduced, distributed, displayed, published or broadcast without the prior permission of the owner of that content. |

